
Stable Isotope Labeled (SIL) Peptides
SIL peptides definition
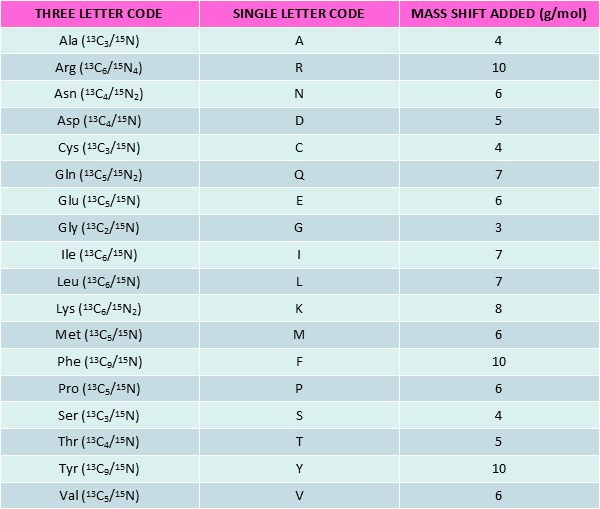
Stable Isotope Labeled peptides (SIL peptides), also known as heavy peptides, are made of heavy amino acids (AA). The latter are derived from natural amino acids by substitution of certain atoms with their heavy isotope variants i.e. 13C, 15N and 2H for 12C, 14N and 1H respectively.
SIL peptides applications
SIL peptides show identical chemical and physical properties as their non-labeled counterparts. However, upon certain conditions, the mass difference makes labeled and unlabeled peptides behave differently. This Δm constitutes the basis for using SIL peptides in quantitative proteomics or Nuclear Magnetic Resonance (NMR) spectroscopy.
SIL peptides for quantitative proteomics
Quantitative proteomics is an analytical technique for quantifying specific proteins in a sample. This quantification is either relative or absolute and SIL peptides are involved in the latter.
Relative protein quantification
Relative protein quantification allows scientists to study proteome expression among different situations. To do so, Isotope Coded Affinity Tags (ICAT) or isobaric Tags for Relative and Absolute Quantification (iTRAQ) can be used.
ICAT is a gel free MS-based technique used to compare up to two samples. Thanks to specific chemical reagents, cysteine containing proteins of each sample are "heavily" (13C isotopic linker) or "lightly" (12C isotopic linker) labeled. After isolation and purification by affinity chromatography, differentially labeled samples are combined in one vial for Liquid Chromatography combined to mass spectrometry (LC/MS) analysis. Relative levels of proteins of interest are then determined by comparing the isotope pairs' peak intensities, reflecting the amount of corresponding proteins in each sample.
In order to quantify all proteins from different sources in a single experiment, iTRAQ were developed. These chemical tagging reagents are made of a unique charged reporter group, a neutral balance group, and a peptide reactive group that binds covalently to the N-terminus and side chain amines of all samples' proteins. After tagging all proteins of each sample with a specific iTRAQ reagent, Liquid Chromatography combined to tandem mass spectrometry (LC-MS/MS) is used to fractionate (LC) and fragmentate (MS/MS) each protein of interest. A database search can then be performed to identify similar labeled peptides, and hence the corresponding proteins, to determine their relative amount by comparison of the peak areas (LC) and the resultant peak ratios for MS/MS reporter ions.
Absolute protein quantification
Even though the vast majority of quantitative protein data are generated using relative quantification, this approach is restricted to a limited number of samples that are prepared as uniformly as possible for the study of one protein in different conditions.
In order to overcome these limitations, Absolute protein QUAntification (AQUA) was developed. Thanks to Selected Reaction Monitoring (SRM) or Multiple Reaction Monitoring (MRM) in a tandem mass spectrometer, the absolute concentration of a protein and its post-translational modifications, such as phosphorylation, can be determined with high selectivity, low background signals and high duty cycle.
In addition, this analytical method is also used for gene silencing studies because the protein supposed to be transcribed doesn't need previous isolation of the native peptide improving greatly the data quality, as well as the number of peptides to be analyzed in a single LC/MS analysis.
Label-free AQUA relies on SIL peptides, used as internal standards, to mimic native tryptic peptides formed after protein's digestion. This method is based on the relationship between the MS signal response and the protein concentration according to which the three most intense tryptic peptides per mole of protein is constant within less than 10% variation.
SIL peptides for Nuclear Magnetic Resonance
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful technique to investigate the structure, dynamics, and molecular interactions of biomolecules in solution. SIL peptides combined with this analytical method reduce the complexity of spectra and allow specific study of proteins' dissociation from their target, as well as protein splicing mechanism, for instance.
Custom enriched isotope peptides
Whether it be for protein structural analysis or quantification, SB-PEPTIDE offers a custom peptide synthesis service dedicated to proteomics. Using Fmoc-based solid-phase peptide synthesis, SIL peptides can be supplied with the following characteristics :
| Specifications | Individual custom SIL peptides | Peptide libraries (96 well plate) |
| Scale | 1 mg, 2 mg, 5 mg and over | 5 µmol/peptide |
| Isotopic enrichment | 13C/15N labeling > 99% | |
| Purity rate | > 80% to > 95% | Crude |
| Aliquoting | Customized (up to 10 µg/tube), delivered in lyophilized or solubilized form | |
| Quality Control | Liquid Chromatography and Mass Spectrometry | |
| Available options | AAA quantification / T,S,Y phosphorylation / Cys CAM / Met[OH] | |
Our 10 days express service is applicable on the products of our Stable Isotope Labeled peptides catalog below, but do not hesitate to contact us to check if your peptides are eligible.
Heavy peptides catalog
| Name | Description | Reference |
| Heavy Angiotensin II |
SIL peptide |
SB037 |
| Heavy Calcitonin peptide | SIL hCT signature peptide quantifier | SB049 |
| Heavy APYTFGQGTK peptide | SIL ADA signature peptide quantifier | SB042 |
| Heavy NYLAWYQQKPGK peptide | SIL ADA signature peptide quantifier | SB287 |
| Heavy SINSATHYAESVK peptide | SIL IFX signature peptide quantifier | SB288 |
| Heavy YASESMSGIPSR peptide | SIL IFX signature peptide qualifier | SB289 |
| Heavy ASGYTFTSYNMHWVK peptide | SIL RTX signature peptide quantifier | SB290 |
| Heavy GLEWIGAIYPGNGDTSYNQK peptide | SIL RTX signature peptide qualifier | SB291 |
| Heavy YASESISGIPSR peptide | SIL CTX signature peptide quantifier | SB292 |
| Heavy ASQSIGTNIHWYQQR peptide | SIL CTX signature peptide qualifier | SB293 |
| Heavy LSITIRPR peptide | SIL DPL signature peptide quantifier | SB294 |
| Heavy DYAMTWVR peptide | SIL DPL signature peptide qualifier | SB295 |
| Heavy SSSTAYMHLK peptide | SIL DNX signature peptide quantifier | SB296 |
| Heavy SLEWIGAIDPYYGGTSYNQK peptide | SIL DNX signature peptide qualifier | SB297 |
| Heavy LEWIGEIDPSESNTNYNQK peptide | SIL VDZ signature peptide | SB298 |
| Heavy SGGSIYNEEFQDR peptide | SIL EMZ signature peptide quantifier | SB299 |
| Heavy QAPGQGLEWMGDINTR peptide | SIL EMZ signature peptide qualifier | SB300 |
| Heavy ADALQAGASQFETSAAK peptide | SIL IFX signature peptide | SB300 |
Bioanalysis. 2020 Sep 25;12(19: 1757-6180. doi: https://doi.org/10.4155/bio-2020-0204
Bottom-up sample preparation for the LC–MS/MS quantification of anti-cancer monoclonal antibodies in bio matrices
Therapeutic monoclonal antibodies (mAbs) are rapidly taking over the treatment of many malignancies, and an astonishing number of mAbs is in development. This causes a high demand for quantification of mAbs in biomatrices both for measuring therapeutic mAb concentrations and to support pharmacokinetics and pharmacodynamics studies. Conventionally, ligand-binding assays are used for these purposes, but LC–MS is gaining popularity. Although intact (top-down) and subunit (middle-down) mAb quantification is reported, signature peptide (bottom-up) quantification is currently most advantageous. This review provides an overview of the reported bottom-up mAb quantification methods in biomatrices as well as general recommendations regarding signature peptide and internal standard selection, reagent use and optimization of digestion in bottom-up quantification methods.
Biol. Chem. 2017 Jan 18;398(5-6):687-699. doi: https://doi.org/10.1515/hsz-2017-0104
Strategies in relative and absolute quantitative mass spectrometry based proteomics
Quantitative mass spectrometry approaches are used for absolute and relative quantification in global proteome studies. To date, relative and absolute quantification techniques are available that differ in quantification accuracy, proteome coverage, complexity and robustness. This review focuses on most common relative or absolute quantification strategies exemplified by three experimental studies. A label-free relative quantification approach was performed for the investigation of the membrane proteome of sensory cilia to the depth of olfactory receptors in Mus musculus. A SILAC-based relative quantification approach was successfully applied for the identification of core components and transient interactors of the peroxisomal importomer in Saccharomyces cerevisiae. Furthermore, AQUA using stable isotopes was exemplified to unraveling the prenylome influenced by novel prenyltransferase inhibitors. Characteristic enrichment and fragmentation strategies for a robust quantification of the prenylome are also summarized.
Chem Rev. 2013 Apr 10;113(4):2343–2394. doi:10.1021/cr3003533
Protein Analysis by Shotgun/Bottom-up Proteomics
Proteins are important because they are the direct bio-functional molecules in the living organisms. The term “proteomics” was coined from merging “protein” and “genomics” in the 1990s.3–4 As a post-genomic discipline, proteomics encompasses efforts to identify and quantify all the proteins of a proteome, including expression, cellular localization, interactions, post-translational modifications (PTMs), and turnover as a function of time, space and cell type, thus making the full investigation of a proteome more challenging than sequencing a genome. There are possibly 100,000 protein forms encoded by the approximate 20,235 genes of the human genome,5 and determining the explicit function of each form will be a challenge.
Bioanalysis. 2012 Jul;4(14):1763-1786. doi: https://doi.org/10.4155/bio.12.126
The use of selected reaction monitoring in quantitative proteomics
Selected reaction monitoring (SRM) has a long history of use in the area of quantitative MS. In recent years, the approach has seen increased application to quantitative proteomics, facilitating multiplexed relative and absolute quantification studies in a variety of organisms. This article discusses SRM, after introducing the context of quantitative proteomics (specifically primarily absolute quantification) where it finds most application, and considers topics such as the theory and advantages of SRM, the selection of peptide surrogates for protein quantification, the design of optimal SRM co-ordinates and the handling of SRM data. A number of published studies are also discussed to demonstrate the impact that SRM has had on the field of quantitative proteomics.
Curr Opin Chem Biol. 2008 Oct;12(5):483–490. doi:10.1016/j.cbpa.2008.07.024
Mass Spectrometry for Proteomics
Mass spectrometry has been widely used to analyze biological samples and has evolved into an indispensable tool for proteomics research. Our desire to understand the proteome has led to new technologies that push the boundary of mass spectrometry capabilities, which in return has allowed mass spectrometry to address an ever-increasing array of biological questions. The recent development of a novel mass spectrometer (Orbitrap) and new dissociation methods such as electron transfer dissociation have made possible exciting new areas of proteomic application. Although bottom-up proteomics (analysis of proteolytic peptide mixtures) remains the workhorse for proteomic analysis, middle- and top-down strategies (analysis of longer peptides and intact proteins, respectively) should allow more complete characterization of protein isoforms and post-translational modifications. Finally, stable isotope labeling strategies have transformed mass spectrometry from merely descriptive to a tool for measuring dynamic changes in protein expression, interaction and modification.
